Birçok çalışma, gelişimsel bozukluk olan Rett sendromunu, MECP2 genindeki genel işlev kaybından kaynaklanan tek bir durum olarak ele alsa da, MIT'deki Picower Öğrenme ve Hafıza Enstitüsü'nden nörobilimcilerin yaptığı yeni bir çalışma, genin iki farklı mutasyonunun laboratuvar kültürlerinde birçok farklı anormallik yarattığını gösteriyor. Dahası, her mutasyonun yarattığı temel farklılıkları düzeltmek için farklı tedaviler gerekiyordu.
Yeni açık erişim çalışmanın kıdemli yazarı Mriganka Sur, Picower Enstitüsü ve Beyin ve Bilişsel Bilimler Bölümü'nde Newton Profesörü. “Bireysel mutasyonlar önemlidir,” diyor. “Bu, tek gen bozukluğu için bile tedaviyi kişiselleştirme yaklaşımıdır.”
Çalışma, her mutasyona sahip Rett sendromu hastalarından bağışlanan deri hücreleri veya kan hücrelerinden türetilen “organoidler” veya “mini beyinler” olarak adlandırılan gelişmiş 3D insan beyin dokusu kültürlerini kullandı. Baş yazar Tatsuya Osaki, Picower Enstitüsü'nde araştırma bilimcisi, organoidlerin her mutasyonun belirli sonuçlarını modelleme yeteneğinin, bilim insanlarının sadece MECP2'yi genel olarak devre dışı bıraktığı önceki çalışmalarda ortaya çıkmamış mutasyon-spesifik içgörüler elde etmesine olanak tanıdığını söylüyor. Organoidler ayrıca, her mutasyonun farklı hücre türlerini ve etkileşimlerini nasıl etkilediğini anlamak için yeni bir fırsat sundu.
Ayrı etkiler
MECP2'deki 800'den fazla mutasyon Rett sendromuna neden olabilir, ancak sadece sekizi vakaların %60'ından fazlasını oluşturuyor. Sur ve Osaki, Rett sendromu vakalarının %7-8'ini temsil eden, yalnızca bir DNA baz çiftindeki (916C>T) farkı içeren R306C mutasyonunu seçti. Seçtikleri diğer mutasyon olan V247X, tek bir DNA baz silinmesi (705Gdel) ile genin protein ürününün üretimini durdurduğu için çok daha nadir ve şiddetlidir; bu da proteinin sadece hatalı değil, aynı zamanda eksik olduğu anlamına gelir.
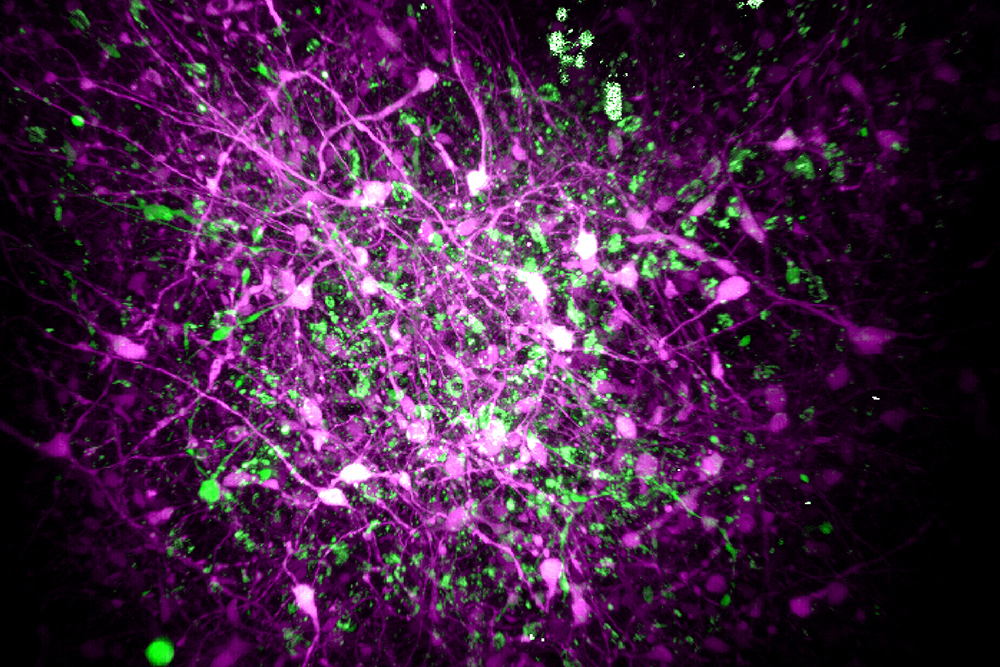
Üç ay boyunca kültürlenen organoidlerde, her mutasyon, kontrol organoidlerine kıyasla bazı ortak ama bazen de farklı sonuçlar üretti. Ekibin birçok deneyinde, organoidlerin yaklaşık 1 milimetre kalınlığında hücresel düzeyde çözünürlük sağlayan “üç-foton” mikroskopları kullandı ve hem yapılarını (“üçüncü harmonik üretim” görüntüleme yoluyla) hem de nöronlarının canlı aktivite desenlerini (kalsiyum floresansı yoluyla) çözümledi.
Örneğin, bilim insanları V247X organoidlerinin kontrollerinden birkaç yapısal farklılık sergilediğini gözlemledi — daha büyüklerdi ve çeşitli katmanların farklı kalınlıklarına sahiptiler — ancak R306C organoidleri kontrollerine çok daha benziyordu. Her iki mutasyonu taşıyan organoidler, kontrol karşılaştırıcılarına kıyasla nöronlarından daha az gelişmiş akson projeksiyonları sergiledi.
Organoidlerdeki sinir aktivitesi ve bağlantı özelliklerini inceleyen bilim insanları, her iki mutasyonda da bazı benzer eksiklikler buldu. Her ikisi de kontrollerine kıyasla azalmış ateşleme aktivitesi ve nöronlar arasındaki senkronizasyon gösterdi.
Ama bilim insanları diğer özelliklere baktıklarında, organoidler birbirinden ayrılmaya başladı. Özellikle, “küçük dünya eğilimi” (SWP) adı verilen ağ yapılarının verimliliğini gösteren bir gösterge, R306C organoidlerinde azalmış, V247X organoidlerinde ise artmıştı. Bu, her iki mutasyonun da bilgi işleme için tipik ağ yapılarını geliştirmeyi değiştirdiği, ancak farklı yönlerde olduğu anlamına geliyor.
Sonuçlarının Rett sendromu hastaları için anlamlı olduğundan emin olmak için ekip, Boston Çocuk Hastanesi'nden Charles Nelson ile işbirliği yaptı; Nelson'un ekibi, farklı Rett mutasyonlarına sahip birkaç çocukta EEG ölçümleri yaptı. Örnek küçük olmasına rağmen, araştırmacılar gönüllülerde organoidlerdeki gibi SWP özelliğinin değiştiğine dair göstergeler ölçtü.
Son olarak, uyarıcı nöronları bir renkte ve inhibitör nöronları farklı bir renkte parlayacak şekilde etiketleyerek, bilim insanları V247X organoidlerinde farklı nöral tür






